Recently, a commenter on the Center for Science and Culture’s Facebook page asked about a paper by the late biochemist Russell F. Doolittle (1931-2019) [1] in relation to Michael Behe’s claim, defended in Darwin’s Black Box [2] (and in this video), that the blood clotting cascade is irreducibly complex. Doolittle claims to show “Step-by-step evolution of vertebrate blood coagulation.” The purpose of this article is to respond to this claim. First, I will provide a brief description of the blood clotting cascade, for the purpose of bringing readers unfamiliar with the pathway up to speed. Second, I will discuss why vertebrate coagulation is considered to be an irreducibly complex system that poses a significant challenge to evolutionary explanations. Third and finally, I will summarize the key points of the Doolittle paper and offer an evaluation of whether it calls Behe’s argument into question. Since Doolittle has also published a book dealing with this subject [3], in which he elaborates on the arguments expressed in the paper in more detail, I will occasionally refer to things said in the book as well.
As we shall see, Doolittle’s proposed scenario does not even touch upon Behe’s thesis, since even in his purported first step in the evolution of vertebrate blood clotting, Doolittle essentially helps himself to all of the components that Behe argued were part of the irreducibly complex core of the system. Thus, Doolittle never even attempts to explain the evolution of the blood clotting system that Behe described as “irreducibly complex.” Doolittle also completely ignores difficulties in adding new factors to the system via unguided evolutionary processes, including the clotting inhibitors that are needed to prevent excessive clot formation, as well as those factors that dismantle clots. This is a glaring omission, since the emergence of the coagulation cascade along the lines proposed by Doolittle would quite probably result in runaway thrombosis, which would entail a significant fitness cost, to say the least. We shall discuss these problems, among others, in the final section of this article.
An Overview of Vertebrate Blood Clotting
The Formation of the Platelet Plug
Blood clotting, also known as coagulation, is a complex physiological process that plays a crucial role in maintaining the integrity of the circulatory system. It involves a series of intricate molecular and cellular interactions that result in the formation of a clot at the site of injury within a blood vessel. This process prevents excessive bleeding while promoting wound healing.
Upon injury, platelets adhere to the exposed collagen fibers in the damaged area. [4] The platelets then undergo a conformational change and release various substances stored within their granules, which promote further platelet activation and attract more platelets to the site of injury, forming a plug over the hole. Activated platelets bind together, forming aggregates or clumps, reinforcing the platelet plug and creating a temporary seal over the injured area. This seal is a short-term solution, however, and is not strong enough to hold for long. As blood continues to flow through the injured vessel, it can dislodge the loosely adhering platelets. Thus, the formation of this platelet plug (and the injury itself) initiates the coagulation cascade.
The Fibrin Gel
The coagulation pathway involves many different components, which can be difficult to keep track of. For ease of following the description that follows, I recommend referring to the figure below, which depicts aspects of the human coagulation system. An arrow from one component to another indicates that the former activates the latter. A barred line signifies inhibition of one protein by another.
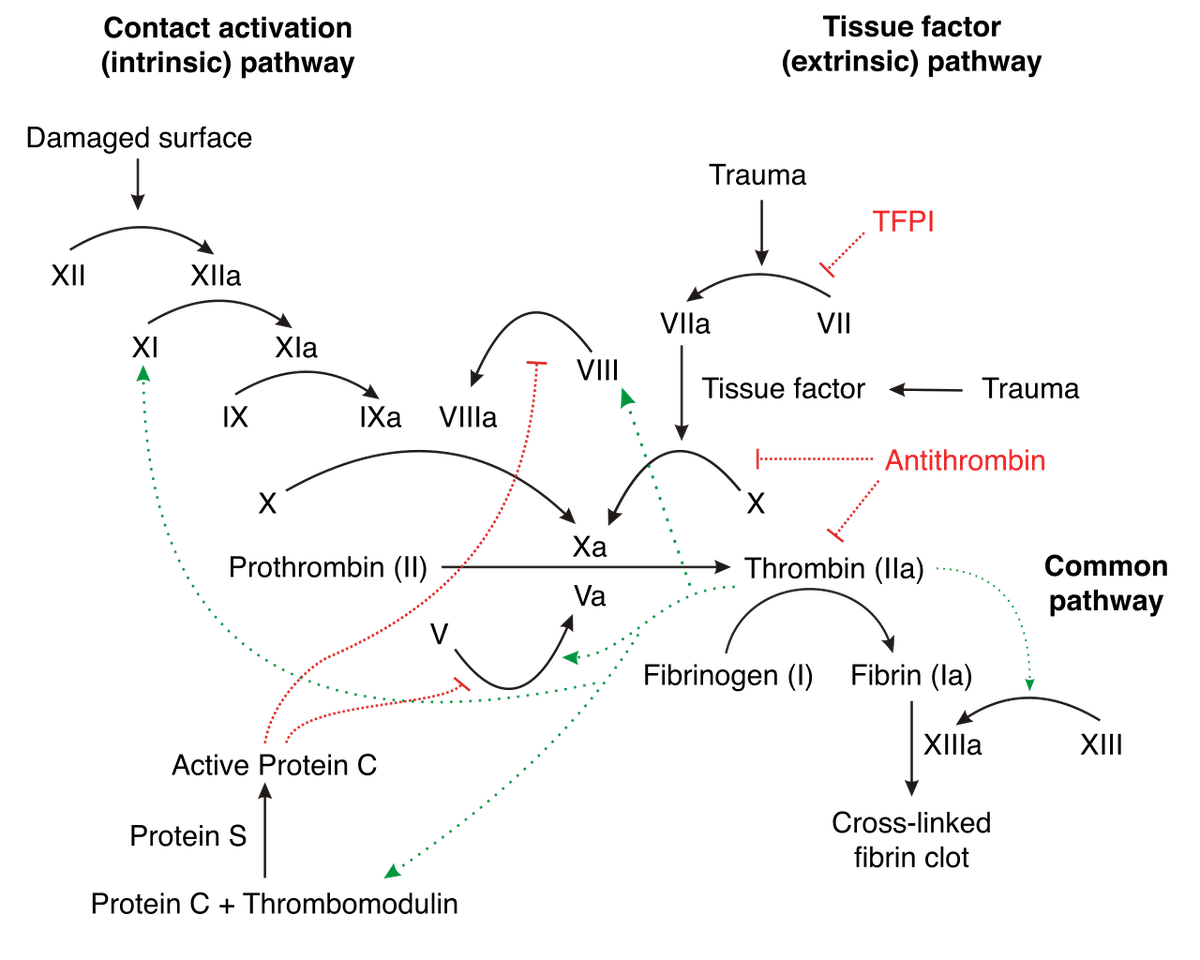
Image credit: Joe D, CC BY-SA 3.0 http://creativecommons.org/licenses/by-sa/3.0/, via Wikimedia Commons.
Vertebrate blood coagulation is best understood by focusing first on the ultimate objective of the cascade, which is the formation of a fibrin gel that reinforces the initial platelet plug, thereby strengthening the clot. The clot itself is made of fibers composed of the protein fibrin, which circulates in an inactive form (fibrinogen) in the blood plasma [5,6], shown in the figure below.
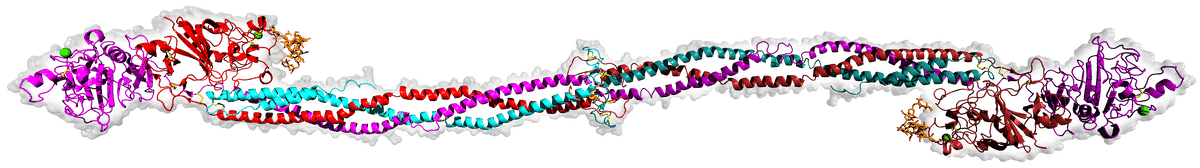
Image source: Wikimedia Commons.
Fibrinogen is comprised of three pairs of polypeptide chains, known as Aα, Bβ, and γ chains, which are held together by disulfide bonds. Fibrinogen becomes activated when another protein, called thrombin, cleaves specific peptide bonds in the fibrinogen molecule, specifically near the N-terminus of the Aα and Bβ chains. [7] This cleavage removes small peptide fragments called fibrinopeptides A and B, respectively, from the fibrinogen molecule, resulting in a similar protein with remarkably different functions referred to as fibrin. The cleavage of fibrinopeptides exposes new binding sites on the fibrin molecule, allowing the individual fibrin molecules to polymerize into a clot. [8] The fibrin molecules further aggregate and form a mesh-like network, which is stabilized by the enzyme factor XIIIa. [9] Factor XIIIa catalyzes the crosslinking of fibrin molecules through the formation of covalent bonds between specific amino acid residues, creating a stable fibrin clot. The resulting fibrin clot, together with the platelets, provides a physical barrier at the site of injury, preventing further blood loss. It also serves as a scaffold for other components of the clotting process, which aggregate on the fibrin network to form a stable blood clot.
If the pathway consisted only of fibrinogen and thrombin, thrombin would constantly cleave fibrinogen, and the consequence would be uncontrolled and excessive clotting throughout the bloodstream. To avoid this, it is essential that the process be carefully regulated. Blood clotting involves the use of proenzymes, which are enzymes that are retained in an inactive state and need to be converted into active enzymes through specific cleavage by proteases such as thrombin.
Thrombin itself exists in an inactive form, prothrombin. To convert prothrombin into active thrombin, another enzyme (factor Xa), along with its cofactor (factor Va), assembles on the surface of platelets or other phospholipid membranes to form the prothrombinase complex. [10] This complex provides the platform for the subsequent activation of prothrombin. The conversion of prothrombin to thrombin, mediated by the prothrombinase complex, occurs (as with fibrinogen) through a proteolytic cleavage of prothrombin at specific sites. Curiously, factor V also exists in an inactive state, but is cleaved and activated (to form factor Va) by thrombin itself (a small trace of which circulates in the bloodstream due to a low rate of cleavage of prothrombin by factor Xa). [11] Once activated, factor Va plays a critical role in the amplification of the coagulation process by enhancing the activity of factor X and promoting the production of more thrombin. This makes the coagulation cascade autocatalytic, since the activation of clotting factors leads to the activation of more of the same proteins.
Factor Xa also exists in an inactive form, factor X. Factor X may be activated by two different pathways: the extrinsic and intrinsic pathway. In the extrinsic pathway, so-named because it is triggered by the external factors, tissue factor (also known as factor III) is released. [12] Tissue factor forms a complex with factor VII, leading to the activation of factor X. The first step of the intrinsic pathway, so-named because all of the components required for its initiation and progression are present within the blood itself, involves the activation of factor XII (also called Hageman factor) by contact with negatively charged surfaces, such as exposed collagen at the site of injury. [13] Activated factor XII then activates factor XI, which, in turn, activates factor IX. Activated factor IX (IXa) forms a complex with its cofactor, factor VIIIa, on a phospholipid surface. This complex, along with calcium ions, is called the tenase complex or intrinsic tenase. [14,15] The tenase complex plays a crucial role in amplifying the clotting process by cleaving, and thereby activating, factor X.
Preventing Excess Clotting
To prevent excess clotting and ensure that the clotting cascade remains localized to the site of injury, there are several regulatory mechanisms. [16] Antithrombin III (ATIII) is a natural anticoagulant that inhibits the activity of thrombin and several other coagulation factors, including factor Xa and factor IXa. It achieves its anticoagulant effects through a mechanism called “serpin” inhibition. [17] Serpins (serine protease inhibitors) are a class of proteins that regulate the activity of proteases, including those involved in blood clotting. ATIII binds to Thrombin’s active site, effectively blocking its ability to cleave fibrinogen into fibrin. By inhibiting thrombin, antithrombin III indirectly helps regulate the activation of factor V and thereby prevents excessive clotting. [18]
Thrombin not only activates factor V but also activates protein C, which, in the presence of its cofactor protein S, inactivates factor Va (activated form of factor V). [19] Protein C cleaves factor Va at specific sites, rendering it less active and inhibiting its procoagulant properties. This negative feedback loop helps to limit and regulate the clotting process. Tissue factor pathway inhibitor (TFPI) is a protein that directly inhibits the activity of factor Xa and the factor VIIa-tissue factor complex. [20] By inhibiting these factors, TFPI also indirectly prevents excessive activation of factor V and, thereby, of thrombin.
Clot Retraction and Dissolution
After the clot is formed, it undergoes retraction, which involves the contraction of fibrin by platelets within the clot, resulting in the clot becoming denser. [21] This process helps to reduce the size of the clot and brings the edges of the wound closer together. Eventually, as the wound heals, the clot needs to be dissolved to restore normal blood flow. Plasmin, a proteolytic enzyme, breaks down the fibrin meshwork into soluble fragments, leading to the dissolution of the clot. [22] Plasmin is generated from plasminogen by tissue plasminogen activator (t-PA) or urokinase-type plasminogen activator (u-Pa), among other molecules.
The Role of Vitamin K
Several of the proteins discussed here depend upon vitamin K for their synthesis — these are prothrombin, factors VII, IX, and X, as well as proteins C and S (i.e., the anticoagulant proteins that serve to inhibit excessive clot formation). Vitamin K is essential for the post-translational modification of these clotting factors. Without adequate vitamin K, these proteins cannot undergo the necessary chemical changes, which would impair their ability to function properly in the coagulation process. For this reason, deficiency in vitamin K can lead to a bleeding disorder known as vitamin K deficiency bleeding, or coagulopathy.
Summary
In summary, vertebrate blood clotting is an incredible, tightly regulated, multi-component cascade that intuitively points to intelligent design. This is in view of its goal-directedness towards its final end, which is producing a successful clot. In the next section, I will discuss why unguided evolutionary explanations are implausible in accounting for the origins of the coagulation pathway, in order to set the stage for my evaluation of a particular attempt to offer an evolutionary account of blood clotting by biochemist Russell Doolittle.
Why the Blood Clotting Cascade Challenges Evolution
What components are essential to the coagulation cascade? If we limit our analysis, for simplicity, to those components that make up the common pathway (i.e., after the convergence of the intrinsic and extrinsic pathways), the essential proteins include fibrinogen, prothrombin, factor X, and factor V. In the absence of fibrinogen (a condition known as afibrinogenemia), the mesh-like network that stabilizes the blood clot will not form. [23] In the absence of prothrombin, the result is a bleeding disorder called hypoprothrombinemia. [24] Since no thrombin is produced, fibrinogen is not converted to fibrin and thus the system fails, again, to form the mesh-like network that is necessary for the formation of a stable clot. In the absence of factor V (known as Owren’s disease), the production of thrombin is significantly reduced, with a similar result. [25] In the absence of factor X (known as Stuart-Prower disease), the production of thrombin is impaired, which again prevents blood clot formation. [26]
Carefully and Finely Tuned
Moreover, as Michael Behe explains in Darwin’s Black Box, the coagulation process needs to be carefully and finely tuned [27]:
If only a small amount of fibrinogen were available it would not cover a wound; if a primitive fibrin formed a random blob instead of a meshwork, it would be unlikely to stop blood flow. If the initial action of antithrombin were too fast, the initial action of thrombin too slow, or the original Stuart factor [i.e., Factor X] or Christmas factor [i.e., Factor IX] or antihemophilic factor [i.e., Factor VIII] bound too loosely or too tightly (or if they bound to the inactive forms of their targets as well as the active forms), then the whole system would crash.
Indeed, “the quality and character of the fibrin clots generated in fish and mammals do not appear to be significantly different.” [28] Thus, the earliest vertebrates apparently formed fibrin clots that were not less effective or durable than those found in humans. How is this to be explained by a gradual trial-and-error process?
Furthermore, once the clotting has begun, there must be a mechanism to prevent excessive clotting and to confine the clot to the site of the injury. Otherwise, the result would be widespread clotting throughout the body’s blood vessels, leading to thrombosis and eventual death. Indeed, “this suppression of activity is very important; there is enough prothrombin in one milliliter of plasma to clot all the fibrinogen in the whole body if the prothrombin were all converted to thrombin.” [29] Thus, the coagulation cascade cannot evolve unless there is simultaneously a mechanism in hand for controlling it to prevent excessive and widespread clotting. Both would have to arise at the same time. Thus, the coagulation cascade is balanced on a knife-edge. Maintaining this delicate balance requires intricate mechanisms, ensuring that clotting occurs when needed to prevent excessive bleeding, while also preventing unnecessary clot formation that could lead to harmful consequences such as thrombosis. Any disruptions or imbalances in these regulatory mechanisms can result in severe bleeding or clotting disorders.
A Proenzyme and an Activating Enzyme
Another difficulty is that, from the very start, any new step that was added to the cascade would need both a proenzyme and an activating enzyme to ensure that the proenzyme is turned on at the appropriate time. As Behe explains, while one might envision some simpler system where there is a direct pathway from factor X to fibrinogen, bypassing thrombin entirely, “If a new protein were inserted into the thrombinless system it would either turn the system on immediately — resulting in rapid death — or it would do nothing, and so have no reason to be selected. Because of the nature of a cascade, a new protein would immediately have to be regulated.” [30] Thus, “since each step necessarily requires several parts, not only is the entire blood-clotting system irreducibly complex, but so is each step in the pathway.” [31] A further problem with the hypothetical thrombinless system, entertained above, is that thrombin is highly effective in cleaving fibrinogen at specific sites, generating fibrin monomers that can polymerize to form a stable clot. Directly cutting fibrinogen with factor X alone would not produce the same reliable and robust fibrin network required for effective clot formation, unless an early form of factor X cut fibrinogen at the same sites that thrombin does.
One might try to explain the origins of new blood clotting factors by postulating successive rounds of gene duplication and divergence (where the duplicate copy would still be under the regulation of the enzyme regulating the original factor). However, a major problem here is that the duplication of a gene would create an excess of the protein that the gene codes for. But excessive expression of clotting factors can disrupt the delicate balance of the coagulation cascade, leading to an increased tendency for the formation of blood clots. To take one example, in humans, 2 to 4.5g/L is considered to be the normal range for the concentration of fibrinogen in blood plasma. [32] Doubling this concentration, which occurs during pregnancy, significantly increases the risk of thrombosis — while halving it creates a risk of bleeding. [33] Thus, the relative concentrations of coagulation factors is very precisely balanced. Upsetting this balance can result in conditions such as deep vein thrombosis, pulmonary embolism, or stroke. To take another example, it has been shown that [34],
Elevated (pro)thrombin levels trigger the formation of densely-packed fibrin clots composed of thin fibrin fibers compared to normal clots. Increased thrombin generation in these individuals also increases activation of the thrombin-activatable fibrinolysis inhibitor (TAFI) in vitro. Activated TAFI downregulates fibrinolysis by cleaving C-terminal lysine residues from fibrin and reducing the number of tPA and plasminogen binding sites on fibrin. It has been suggested that the combination of abnormal structure and increased TAFI activation reduces the rate of fibrinolysis and contributes to the increased risk of thrombosis in these individuals.
Thus, a duplicate blood clotting factor gene is likely to be deleterious and purged by purifying selection rather than preserved. Regarding this problem, one might retort that a gene duplication event that knocks the system out of balance might be compensated for by a second gene duplication event that creates a new activating enzyme, thereby bringing the system back into balance. However, among eukaryotic organisms, any particular gene has only a probability of being duplicated, over the span of one million years, of 0.01, “with rates in different species ranging from about 0.02 down to 0.002.” [35] Moreover, “the vast majority of gene duplicates are silenced within a few million years, with the few survivors subsequently experiencing strong purifying selection.” [36] Given the selection costs of carrying a duplicated gene, upsetting the delicate balance of the coagulation cascade, it is unlikely that it would be retained long enough for the appropriate gene to also be duplicated, restoring the balance of the system.
A Challenge to Evolutionary Mechanisms
To conclude this section, the intricacies of vertebrate blood clotting represent a significant challenge to evolutionary mechanisms. The process of clot formation is itself irreducibly complex and must also emerge simultaneously with a mechanism to prevent excessive clotting and to confine the clot to the site of injury. From a neo-Darwinian perspective, it is difficult to envision such a system emerging one step at a time without passing through maladaptive intermediate stages. On the other hand, a complex integration of parts contributing towards a higher-level objective, such as we see associated with coagulation, is precisely what we might expect on a hypothesis of design. In the remainder of this article, I will review Russell Doolittle’s attempt to provide an evolutionary explanation for this pathway.
Has Russell Doolittle Provided an Evolutionary Explanation of the Blood Clotting Cascade?
With the foregoing challenges in mind, let us inspect Russell Doolittle’s paper on the evolution of vertebrate blood clotting [37] to determine to what extent his analysis assuages these concerns. Since Doolittle has also published a book dealing with this subject [38], in which he elaborates on the arguments expressed in the paper in more detail, I occasionally will refer to things said in the book as well.
Do Gene Duplications Explain Vertebrate Blood Clotting?
Doolittle contends that “Many of the proteins involved [in coagulation] are clearly related to one another by gene duplications, and in the past, sequence-based phylogenies have offered insights into the relative order in which certain factors appeared.” [39] But sequence similarity does not necessarily imply common ancestry (due to the possibility of common design) and common ancestry does not necessarily imply a stepwise evolutionary pathway. Michael Behe explains this point [40]:
Although useful for determining lines of descent… comparing sequences cannot show how a complex biochemical system achieved its function—the question that most concerns us in this book. By way of analogy, the instruction manuals for two different models of computer put out by the same company might have many identical words, sentences, and even paragraphs, suggesting a common ancestry (perhaps the same author wrote both manuals), but comparing the sequences of letters in the instruction manuals will never tell us if a computer can be produced step-by-step starting from a typewriter… Like the sequence analysts, I believe the evidence strongly supports common descent. But the root question remains unanswered: What has caused complex systems to form?
This aside, however, Doolittle’s proposed mechanism runs into the problem enumerated above — namely, that duplicating a gene coding for one of the blood clotting factors would lead to that factor becoming over-expressed, disrupting the cascade’s delicate balance and leading to excessive clotting. This difficulty is nowhere even acknowledged in Doolittle’s paper, let alone addressed.
Doolittle raises a valid concern: “The question may be asked, how can new factors be introduced into an existing pathway?” Good question. How does Doolittle respond? He writes, “It was long ago suggested that in the case of clotting pathways, new factors that are the products of gene duplications could easily be sandwiched into the middle of pathways where they initially were only performing the same operation as the original gene product. Only a few amino acid replacements were likely needed to broaden the proteolytic specificity to the point where the duplicon could itself activate the other surviving gene product.” [41] In support of this thesis, Doolittle notes that “all of the vitamin-K dependent proteases (prothrombin, factors VII, IX, and X, and protein C) cleave after arginine residues in the same general regions of their homologous substrates.” [42] This, again, however, runs into the problem described above — namely, that gene duplication events would be likely to upset the delicate balance of the system, increasing the risk of thrombosis. Such duplicate genes are thus unlikely to be preserved by selection. Moreover, a few specific amino acid replacements in animals such as vertebrates (assuming none of them are beneficial until all have arisen) are unlikely to happen on a realistic timescale. As has been much discussed in the academic literature, evolution depends on prohibitively long times to attain and fix multiple co-dependent mutations, where none of them confer a fitness benefit until all have arisen. [43,44,45,46,47,48]. Doolittle gives a time window for the emergence of the coagulation cascade of approximately fifty to a hundred million years, since fibrin clots have never been detected in any protochordate (i.e., organisms lacking a backbone but possessing a notochord at some stage during development) though it has been identified in the earliest vertebrates — that is, jawless fish. Thus, he argues, blood clotting must have arisen between the emergence of protochordates and jawless fish. As he writes in his book [49],
Because fibrin clots have never been observed in our nearest non-vertebrate relatives, the protochordates, we must accept that the clotting system was assembled in the relatively brief interval since protochordates diverged from the lineage leading to vertebrates and the appearance of creatures like the hagfish and lamprey. In years, the available time is estimated to have been 50 to 100 million.
Given that it is highly probable that each step in the evolution of coagulation would require multiple co-dependent mutations (since each proenzyme would have to evolve in a coordinated way with its activating enzyme), this time window appears to be quite brief. Compounding this is the fact that the mutations in the evolving gene duplicate would need to occur in a coordinated way with its own activating enzyme to ensure that the new factor is active only when needed.
Simpler Blood Clotting Systems in Jawless Fish
There are two extant genera of jawless fish — hagfish (pictured above) and lamprey. Doolittle predicts, from an evolutionary framework, that jawless vertebrates would have a simpler blood clotting cascade than the human system described above. Doolittle limits the scope of his analysis to the lamprey, for which there was better genomic data than for hagfish. Doolittle’s research has revealed that lampreys lack both factor IX and factor VIII. [50,51] It may thus be concluded that factors IX and VIII are not essential, at least in the jawless vertebrates. Why are jawless vertebrates not hemophilic, unlike humans who lack these factors? Without knowing more about how coagulation works in jawless vertebrates, it is impossible to say for sure. Clearly, there are other differences as well — since apparently there are two factor X genes in lamprey. Is it possible that one of those is functioning as a factor IX gene? There are also three factors VII. Unfortunately, the precise mechanisms of coagulation in lampreys have yet to be elucidated. Given that humans who are deficient in factors IX or VIII suffer from hemophilia, this provides an indirect justification for believing that there are, quite probably, compensating mechanisms in lampreys that have yet to be uncovered.
Failing to Address Behe’s Argument
A crucial point is that, even if Doolittle’s entirely evolutionary scheme were correct, it would not even address — much less refute — Behe’s original thesis about the blood clotting cascade. In Darwin’s Black Box, Behe only argued that the common pathway is irreducibly complex — he does not give an assessment of whether the intrinsic pathway (to which these factors IX and VIII, missing in jawless fish, belong) or the extrinsic pathway are irreducibly complex as well. [50] To summarize, in Darwin’s Black Box, Michael Behe only argued for irreducible complexity of the common pathway — i.e., the pathway after the convergence of the extrinsic and intrinsic initiation pathways, or what Behe calls the components “after the fork.” Behe made this very explicit in his book, where he wrote [52]:
Leaving aside the system before the fork in the pathway, where some details are less well known, the blood-clotting system fits the definition of irreducible complexity.
The system “before the fork in the pathway” is the intrinsic and extrinsic initiation pathways highlighted by Doolittle. But Behe explicitly leaves this part of the system “aside” and does not argue it is irreducibly complex. Thus, even if Doolittle’s arguments held merit, they would still not refute, even address, the portion of the pathway that Behe argues is irreducibly complex.
Regarding the systems “after the fork,” to my knowledge, there are no vertebrates with a functional coagulation system that lack thrombin, fibrinogen, factor X, or factor V. Clearly, there also has to be some way of activating factor V in response to tissue damage. Thus, to be conservative, blood coagulation must require a minimum of five parts (and probably more) – the very components which Behe argues comprise the irreducibly compelx portion of the blood clotting cascade. Behe’s argument has not been refuted. This same mistake was made in response to Behe by Kenneth Miller, as has been previously noted at Evolution News by Casey Luskin.
Co-Option of Thrombin / Fibrinogen
Doolittle concedes that “it seems unlikely that thrombin and fibrinogen would appear simultaneously,” and posits that “one already existed with an alternative function.” [53] He suggests that “fibrinogen may have had a role in cell-cell interactions, a property of many proteins with fibrinogen-related domains.” [54] Doolittle postulates that “a more likely scenario, however, is that thrombin had an early role in agglutinating thrombocytes by proteolyzing cell surface proteins, something it is known to do today, attacking a set of G-protein-coupled receptors called PAR proteins.” [55] On this hypothesis, “a tissue factor would become exposed during the course of injury, activating prothrombin that would then clump cells which were the ancient ancestors of mammalian platelets. A GLA domain could have helped to keep thrombin localized on the surface of the thrombocytes.” [56]. The emergence of fibrinogen “would allow thrombin to broaden its attack, generating a more durable clot composed of fibrin. Duplications of the prothrombin gene would lead to the appearance of factors VII, X, and eventually IX.” [57]
Such a scenario, however, presents a number of problems. One is acknowledged by Doolittle himself: “Besides the GLA domain, thrombin has two kringle domains, usually thought to have an affinity for fibrin. The kinds of domains that interact with tissue factor, however, are the EGF domains found in factors VII, X, and IX.” [58] He, therefore, has to postulate that “there was much domain shuffling in the early stages and that thrombin originally had EGF domains, or no peripheral domains at all.” [59] Second, he makes no attempt to estimate how durable a clot composed of nothing more than clumped cells would be — though, if the initial flow rate was relatively low, this issue could be less of a concern. Finally, a duplication of a prothrombin gene would result in a second prothrombin gene. Doolittle makes no attempt to determine how difficult it would be for factors VII, X and IX to arise from a duplicated prothrombin gene (and evolve in a coordinated way with their corresponding activating enzymes), nor how the overexpression problem described above may be overcome.
Doolittle’s Four Stage Scenario
In his book, Doolittle proposes four stages in the evolution of vertebrate blood clotting, based on the presence or absence of coagulation factors in various animals. The timepoints along evolutionary history at which each of his four stages takes place is illustrated in the diagram below, reprinted under fair use from figure 13.1 of his book. [60]
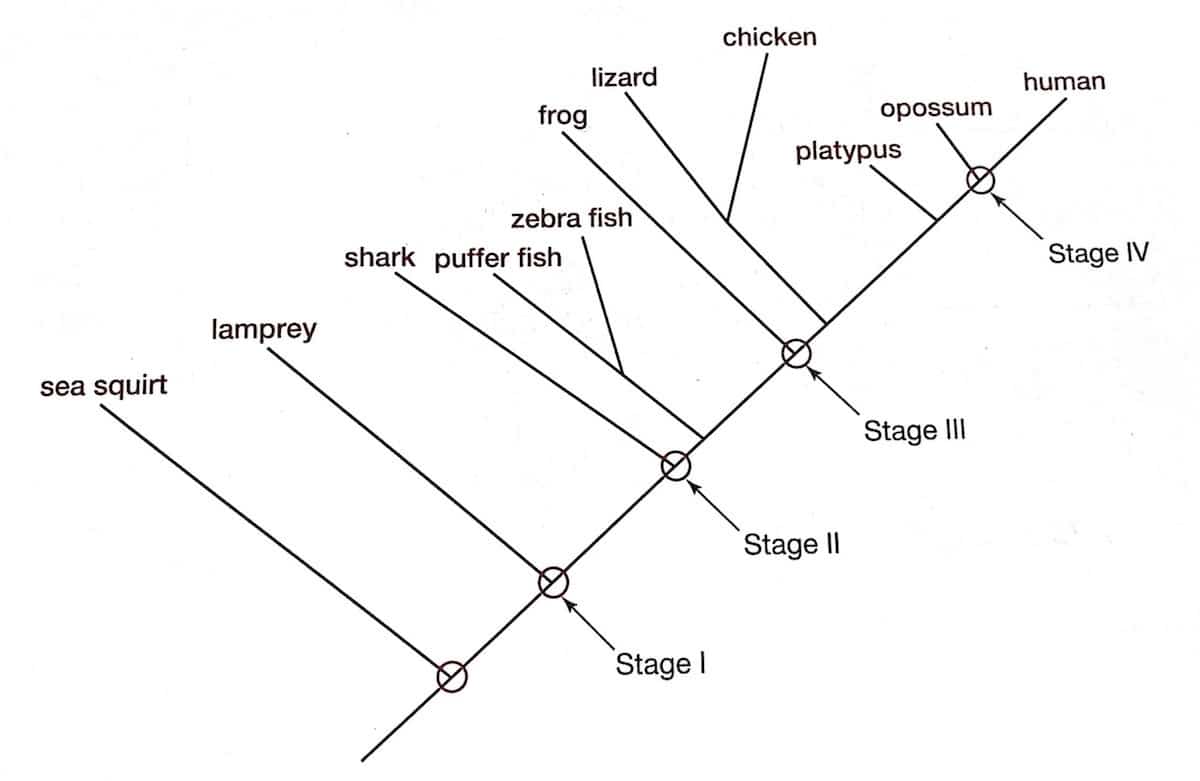
The first stage, according to Doolittle, “existed in the last common ancestor of jawless and jawed vertebrates and was characterized by the presence of only six different proteins, three of which are vitamin K-dependent proteases.” [61] These six proteins included tissue factor, factor VII, factor X, factor V, prothrombin and fibrinogen. The second stage, Doolittle suggests, involved the emergence of factors VIII and IX prior to the evolution of jawed fish. The third stage was characterized by the acquisition of prekallikrein and factor XII. A duplication of the gene coding for prekallikrein, resulting in the origins of factor XI, led, according to Doolittle’s scenario, to the fourth stage.
Notice that even the primitive system that constitutes stage one contains all of the four components that Behe claimed to comprise the irreducibly complex system — that is, thrombin, fibrinogen, factor X and factor V — in addition to tissue factor and factor VII (key components of the extrinsic pathway, required for initiating coagulation in response to tissue damage). Behe’s hypothesis predicts that every coagulation system will contain these four proteins (or equivalents), in addition to there being some way of activating factor V in response to tissue damage. This is precisely what the data show. The only aspect of the coagulation cascade that emerged subsequent to the origins of coagulation itself, then, so far as can be told by the data, are the components that make up the intrinsic pathway. That the intrinsic pathway is redundant is not particularly surprising, since there only needs to be one mechanism by which factor V is activated. Adding the intrinsic pathway is certainly helpful, though not necessarily essential. The intrinsic pathway serves to amplify the coagulation response initiated by the extrinsic pathway, and provides additional layers of activation and enzyme generation. Again, Behe’s fundamental thesis about irreducible complexity of the blood clotting cascade has not been refuted or even touched by Doolittle’s arguments.
But perhaps the most damning problem confronting Doolittle’s proposed scenario is that his analysis entirely ignores all of the clotting inhibitors (such as antithrombin, protein C, and protein S) that prevent excessive clot formation, as well as those factors that dismantle clots (as he himself acknowledges [62]). And this even though he correctly notes elsewhere that “this suppression of activity is very important; there is enough prothrombin in one milliliter of plasma to clot all the fibrinogen in the whole body if the prothrombin were all converted to thrombin.” [63] Thus, the emergence of the pathway for coagulation along the lines suggested by Doolittle would likely result in runaway thrombosis without the simultaneous advent of inhibitors.
A Formidable Challenge
In summary, the coagulation cascade presents a formidable challenge to evolutionary theory. While Doolittle is to be commended for his work in attempting to provide an evolutionary account for coagulation, the failure of those attempts underscores the difficulty of the problem. They simply do not address the issues at the heart of Behe’s argument in Darwin’s Black Box, which remains untouched by Doolittle’s thesis.
Footnotes
[1] Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35-40.
[2] Behe, MJ. Darwin’s Black Box: The Biochemical Challenge to Evolution. Free Press 1996.
[3] Doolittle RF. The Evolution of Vertebrate Blood Clotting. University Science Books 2013.
[4] Periayah MH, Halim AS, Mat Saad AZ. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int J Hematol Oncol Stem Cell Res. 2017 Oct 1;11(4):319-327.
[5] Kattula S, Byrnes JR, Wolberg AS. Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arterioscler Thromb Vasc Biol. 2017 Mar;37(3):e13-e21.
[6] Pieters M, Wolberg AS. Fibrinogen and fibrin: An illustrated review. Res Pract Thromb Haemost. 2019 Mar 4;3(2):161-172.
[7] Greenberg CS, Miraglia CC, Rickles FR, Shuman MA. Cleavage of blood coagulation factor XIII and fibrinogen by thrombin during in vitro clotting. J Clin Invest. 1985 May;75(5):1463-70.
[8] Weisel JW, Litvinov RI. Fibrin Formation, Structure and Properties. Subcell Biochem. 2017;82:405-456. doi: 10.1007/978-3-319-49674-0_13.
[9] Lorand L. Factor XIII and the clotting of fibrinogen: from basic research to medicine. J Thromb Haemost. 2005 Jul;3(7):1337-48.
[10] Krishnaswamy S. The transition of prothrombin to thrombin. J Thromb Haemost. 2013 Jun;11 Suppl 1(0 1):265-76.
[11] Keller FG, Ortel TL, Quinn-Allen MA, Kane WH. Thrombin-catalyzed activation of recombinant human factor V. Biochemistry. 1995 Mar 28;34(12):4118-24.
[12] Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007 Aug;27(8):1687-93.
[13] Grover SP, Mackman N. Intrinsic Pathway of Coagulation and Thrombosis. Arterioscler Thromb Vasc Biol. 2019 Mar;39(3):331-338.
[14] Autin L, Miteva MA, Lee WH, Mertens K, Radtke KP, Villoutreix BO. Molecular models of the procoagulant factor VIIIa-factor IXa complex. J Thromb Haemost. 2005 Sep;3(9):2044-56.
[15] Childers KC, Peters SC, Lollar P, Spencer HT, Doering CB, Spiegel PC. SAXS analysis of the intrinsic tenase complex bound to a lipid nanodisc highlights intermolecular contacts between factors VIIIa/IXa. Blood Adv. 2022 Jun 14;6(11):3240-3254.
[16] Dahlbäck B. Blood coagulation and its regulation by anticoagulant pathways: genetic pathogenesis of bleeding and thrombotic diseases. J Intern Med. 2005 Mar;257(3):209-23.
[17] Sanrattana W, Maas C, de Maat S. SERPINs-From Trap to Treatment. Front Med (Lausanne). 2019 Feb 12;6:25.
[18] van Boven HH, Lane DA. Antithrombin and its inherited deficiency states. Semin Hematol. 1997 Jul;34(3):188-204.
[19] Kalafatis M, Mann KG. Role of the membrane in the inactivation of factor Va by activated protein C. J Biol Chem. 1993 Dec 25;268(36):27246-57. PMID: 8262965.
[20] Wood JP, Ellery PE, Maroney SA, Mast AE. Biology of tissue factor pathway inhibitor. Blood. 2014 May 8;123(19):2934-43.
[21] Tucker KL, Sage T, Gibbins JM. Clot retraction. Methods Mol Biol. 2012;788:101-7.
[22] Alkjaersig N, Fletcher AP, Sherry S. The mechanism of clot dissolution by plasmin. J Clin Invest. 1959 Jul;38(7):1086-95.
[23] Simurda T, Asselta R, Zolkova J, Brunclikova M, Dobrotova M, Kolkova Z, Loderer D, Skornova I, Hudecek J, Lasabova Z, Stasko J, Kubisz P. Congenital Afibrinogenemia and Hypofibrinogenemia: Laboratory and Genetic Testing in Rare Bleeding Disorders with Life-Threatening Clinical Manifestations and Challenging Management. Diagnostics (Basel). 2021 Nov 19;11(11):2140.
[24] Mazodier K, Arnaud L, Mathian A, Costedoat-Chalumeau N, Haroche J, Frances C, Harlé JR, Pernod G, Lespessailles E, Gaudin P, Charlanne H, Hachulla E, Niaudet P, Piette JC, Amoura Z. Lupus anticoagulant-hypoprothrombinemia syndrome: report of 8 cases and review of the literature. Medicine (Baltimore). 2012 Sep;91(5):251-260.
[25] Ehtisham M, Shafiq MA, Shafique M, Mumtaz H, Shahzad MN. Owren’s Disease: A Rare Deficiency. Cureus. 2021 Aug 10;13(8):e17047.
26] Chatterjee T, Philip J, Nair V, Mallhi RS, Sharma H, Ganguly P, Biswas AK. Inherited Factor X (Stuart-Prower Factor) deficiency and its management. Med J Armed Forces India. 2015 Jul;71(Suppl 1):S184-6.
[27] Behe, MJ. Darwin’s Black Box: The Biochemical Challenge to Evolution. Free Press 1996, kindle.
[28] Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35-40.
[29] Ibid., 17.
[30] Behe, MJ. Darwin’s Black Box: The Biochemical Challenge to Evolution. Free Press 1996, kindle.
[31] Ibid.
[32] Grottke O, Mallaiah S, Karkouti K, Saner F, Haas T. Fibrinogen Supplementation and Its Indications. Semin Thromb Hemost. 2020 Feb;46(1):38-49.
[33] Ibid.
[34] Wolberg AS, Campbell RA. Thrombin generation, fibrin clot formation and hemostasis. Transfus Apher Sci. 2008 Feb;38(1):15-23.
[35] Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000 Nov 10;290(5494):1151-5.
[36] Ibid.
[37] Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35-40.
[38] Doolittle RF. The Evolution of Vertebrate Blood Clotting. University Science Books 2013.
[39] Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35-40.
[40] Behe, Michael J. (1996). Darwin’s Black Box: The Biochemical Challenge to Evolution. Free Press, kindle.
[41] Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35-40.
[42] Ibid.
[43] Hössjer O, Bechly G, Gauger A. On the waiting time until coordinated mutations get fixed in regulatory sequences. J Theor Biol. 2021 Sep 7;524:110657.
[44] Sanford J, Brewer W, Smith F, Baumgardner J. The waiting time problem in a model hominin population. Theor Biol Med Model. 2015 Sep 17;12:18.
[45] Durrett R, Schmidt D. Waiting for two mutations: with applications to regulatory sequence evolution and the limits of Darwinian evolution. Genetics. 2008 Nov;180(3):1501-9. Erratum in: Genetics. 2009 Feb;181(2):819-20; author reply 821-2.
[46] Behe MJ, The Edge of Evolution: The Search for the Limits of Darwinism. Free Press 2007.
[47] Behe MJ, Snoke DW. Simulating evolution by gene duplication of protein features that require multiple amino acid residues. Protein Sci.2004 Oct;13(10):2651-64. doi: 10.1110/ps.04802904. Epub 2004 Aug 31.
[48] Axe DD. The Limits of Complex Adaptation: An Analysis Based on a Simple Model of Structured Bacterial Populations. Bio-Complexity 2010.
[49] Doolittle RF. The Evolution of Vertebrate Blood Clotting. University Science Books 2013, 184.
[50] Doolittle RF, Jiang Y, Nand J. Genomic evidence for a simpler clotting scheme in jawless vertebrates. J Mol Evol. 2008 Feb;66(2):185-96. doi: 10.1007/s00239-008-9074-8.
[51] Doolittle RF. Bioinformatic Characterization of Genes and Proteins Involved in Blood Clotting in Lampreys. J Mol Evol. 2015 Oct;81(3-4):121-30.
[52] Behe, Michael J. Darwin’s Black Box: The Biochemical Challenge to Evolution. Free Press 1996, kindle.
[53] Ibid.
[54] Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35-40.
[55] Ibid.
[56] Ibid.
[57] Ibid.
[58] Ibid.
[59] Ibid.
[60] Ibid.
[61] Doolittle RF. The Evolution of Vertebrate Blood Clotting. University Science Books 2013, 185.
[62] Ibid.
[63] Doolittle RF. The Evolution of Vertebrate Blood Clotting. University Science Books 2013, 196.
[64] Ibid., 17.
Note: This article was originally published in August 2023 as a three-part series at Evolution News & Science Today (here, here, and here).